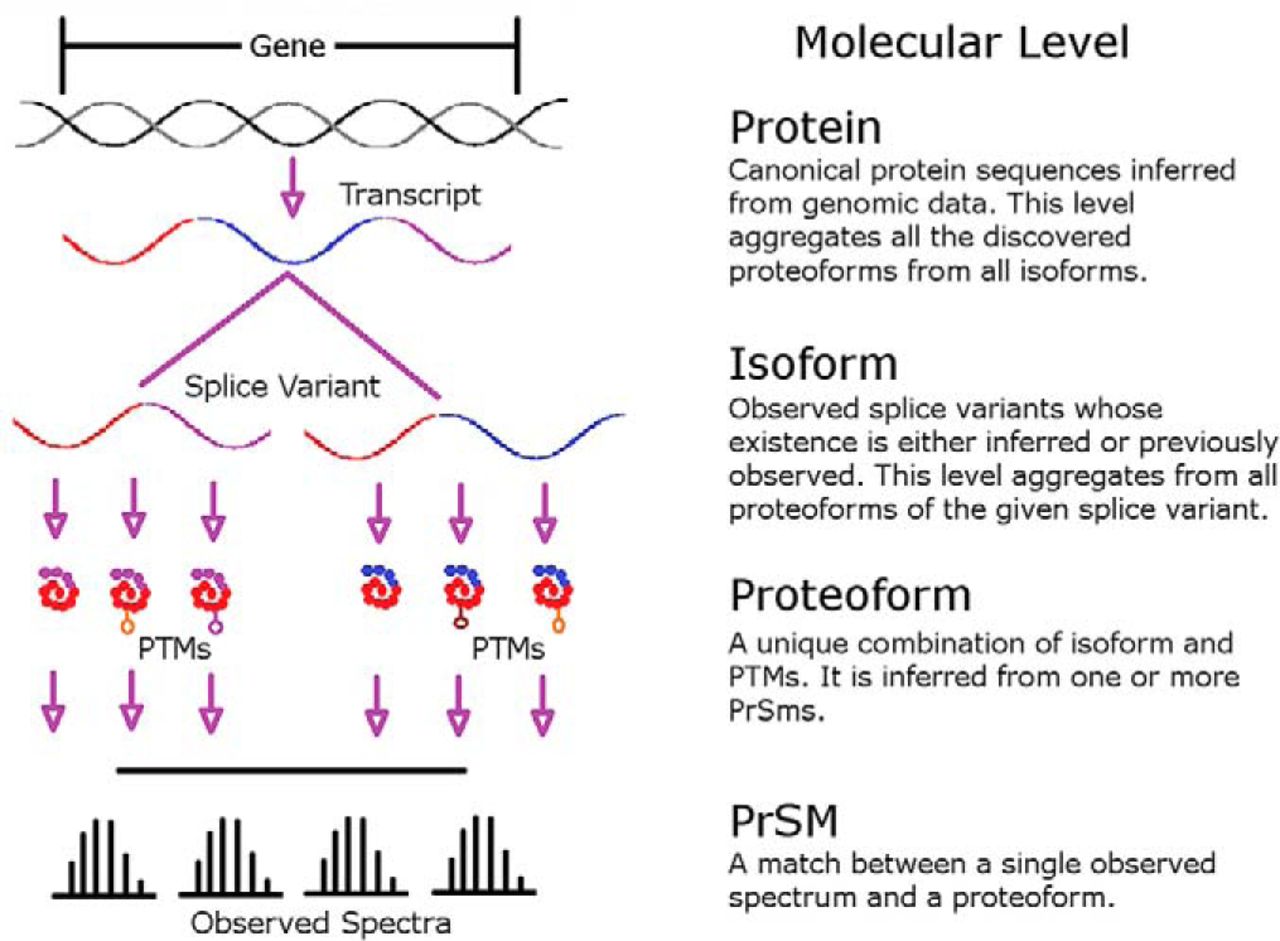
IGNORE MY WRITING. Make beautiful MS/MS spectra easily online by pushing this hideous big button.
Lookin’ to make beautiful spectra for your poster or publication? Just push that big button!!
This might, yet again, be a post mostly for me, because I can’t seem to remember the name of this tool and I keep going to Google Scholar and looking through 2019 papers from the Coon lab. And that isn’t exactly a one-paper-a-year sort of lab. And since I’m already typing I’m going to show you how to use this awesome tool.
You can read about in in MCP here.
If you need to do something fancy, beyond what the online IPSA tool can do, you can download the whole thing on Github here and manipulate it (or run it locally in your own web browser on your offline computer.
I’m going to go through this from a Proteome Discoverer centric application using IMP-PD 2.1 (the free PD version that you can get here.). Sweet! Here is a great tutorial for installing it, I’d not happened into before…as well as a tool I haven’t checked out yet!
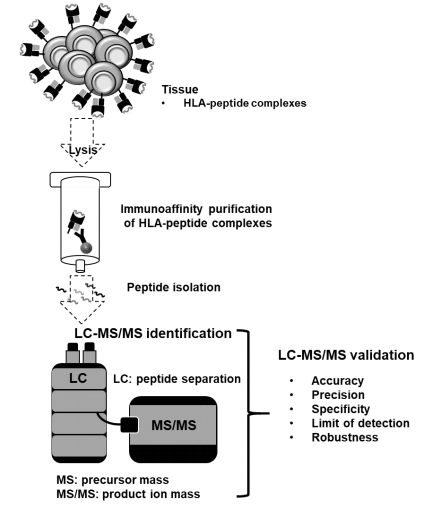
One thing Proteome Discoverer has never done (and — honestly — most, if not all software packages) is made images that your editor and reviewers won’t make fun of. There are some people out there with the kind of exploited students free time that have made them remake all sorts of spectra in things like Adobe illustrator that puts anyone who can’t afford the financial or time costs at a disadvantage. Illustrator images can be so pretty you can hardly look at them
Compare that to your output from your normal tool of choice. Functional? Yes. Pretty? Probably not (and the ones that are pretty, like Scaffold, often don’t contain all the information you want.
IPSA fixes that! You’ll need a couple of things first.
1) Your spectra sequence
2) Exact mass of your post-translational modification, if applicable
3) A clean spectra to work off of
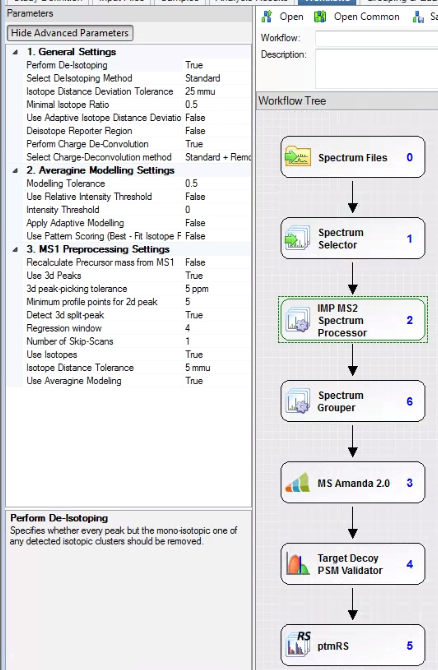
I’ll assume you have both 1 and 2. For number 3 I’m going to use the free version of Proteome Discoverer and version 2.1 because of two nodes that are compatible with 2.1. The IMP-MS2 Spectrum Processor and the Spectrum grouper.
I think the IMP-MS2 spectrum processor has been integrated into MSAmanda 2.0 for later versions of PD, but this is how I’m doing things (PD 2.1, last I checked, was compatible with the largest group of second party nodes and I’ll always keep a version or two installed on everything just in case I need something neat that I don’t have in later versions. I strongly advise you take the 15 minutes when you get a new computer and do the same!)
The MS2 spectrum processor will deconvolute your MS/MS fragments to all singly charged. BOOM! much simpler spectra. It won’t work well, or at all(?) with low resolution spectra, but it works perfectly on the higher resolution ones. You can also deisotope. I do, just so it isn’t so hard to look at everything. It reduces your spectra to the monoisotopic alone.
The spectrum grouper finds your MS/MS spectra (if you have them) that are clearly duplicates, even if you fragmented the +2 and +3 versions and puts them together if you select grouping on “singly charged mass.” To be perfectly honest, I’m not 100% sure I know what this is doing. I thought I did, but I can’t be 100% sure I know what “grouping” means in this context. Meh. I’ll investigate later.
If you’re trying to annotate PTM MS/MS spectra, befinitely throw in the ptmRS/AScore (at least similar enough to consider together) for into the pipeline.
Run this and make some stupendous identifications!
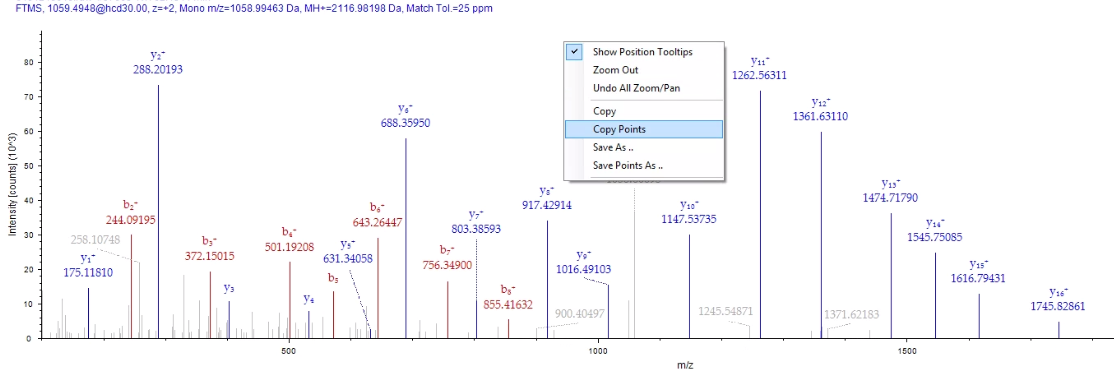
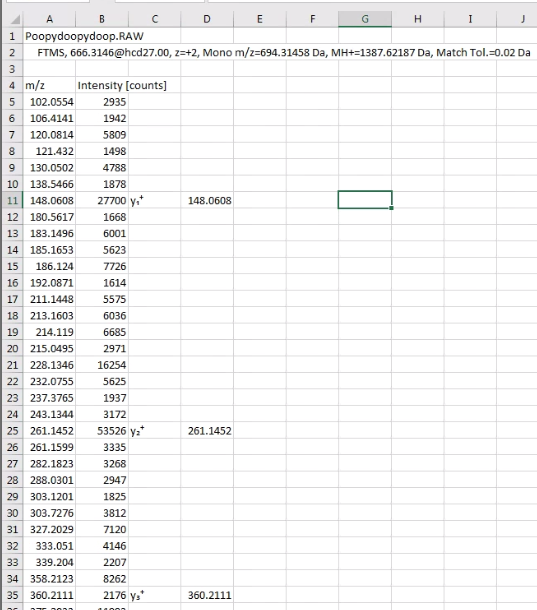
From the PSM tab in PD you can double click on your peptide of interest and bring up a nice and informative MS/MS spectra that your reviewers will make fun of. If you right click on it, you can select “Copy Points”. This will remove all the MS/MS fragments and these annotations and make a 3 column text output.
Next, go to Excel or your Excel like program of choice and paste the data into it. It’ll look something like this. (Please note, examples don’t match in image above/below, because I’m lazy.)
I did this so I could ditch the scan headers, which will confuse IPSA.
All IPSA wants is the MS/MS fragments and intensities. I highlight the cells in rows A &B (columns? I’ll live my entire life without ever truly knowing which is which — thanks, dyslexia, you’re the best!) and copy with ctrl+C, or whatever you Macintosh people use.
Hit the big button at the top of this blog post and go to IPSA.
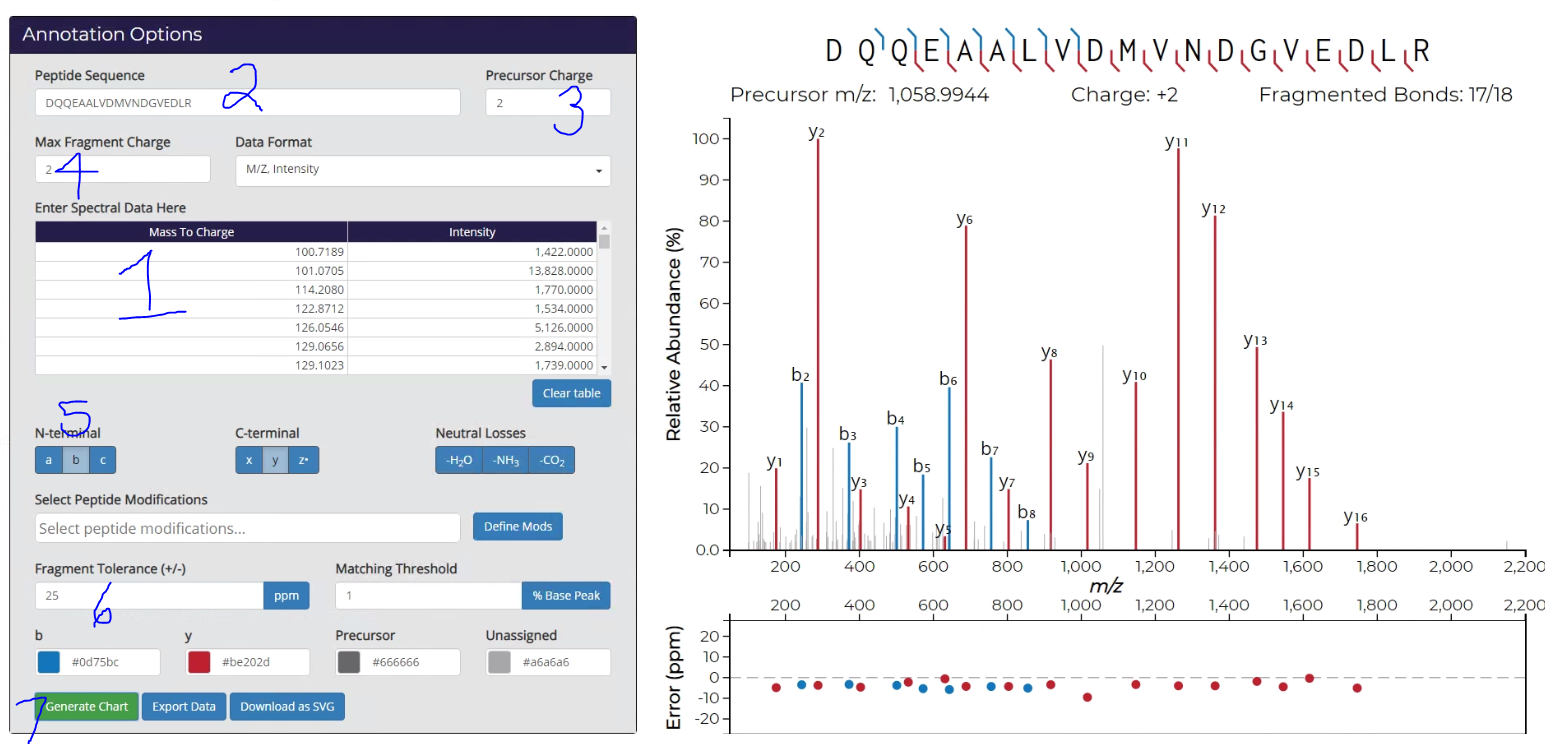
Click to expand the image below. This tool is amazingly straight-forward, but I’m still going to number things.
1) Ctrl+V your cells into the big obvious box!
2) Copy/Paste your peptide sequence in. In PD 2.1 it is easiest to do this from the very top line of the Peptide Summary (where you found the “Copy Points” button a couple of images up from this one.
3) Put in the charge state of your peptide.
4) Determine the charge states of the fragments you want to see. I’ve, in error, selected 2 here, which doesn’t make sense in this workflow, but it did make sense with a low resolution spectra I couldn’t deconvolute. Keep in mind that the IMP tool isn’t perfect and may not catch every MS/MS, if the charge state is particularly high. Worth toying with if you’ve got an unexplained fragment.
5) Select the fragment ions you want to see, and whether you want to see neutral losses. I only use them if there are a lot of unmatched MS/MS spectra.
6) Use a reasonable fragment tolerance in Da or PPM. Matching tolerance is how low vs. base peak you want to still label. It defaults to zero which might be too messy. Putting a 5 in means that you won’t label stuff below 5% of base peak.
Okay — so this is reeeeeaaaly cool. If you don’t like where your labels are, height wise, you can move them. See that y5 for example? Just click and drag it up so you can clearly read it. Then when you….
7) Generate your SVG, it keeps it that way! I’d also recommend exporting the data so that you have this output. It makes a handy CSV with the same title (your peptide sequence) as the SVG and saves it in the same place.
Maybe you’re done! However, if you want to make changes, NOW it’s time to break out Powerpoint or Illustrator.
Illustrator will directly import the SVG and allow you to manipulate it if you know how to use it right. (I don’t and I made my figures much worse). Powerpoint (at least my 365 version) will directly import the SVG. And then I can make changes to it.
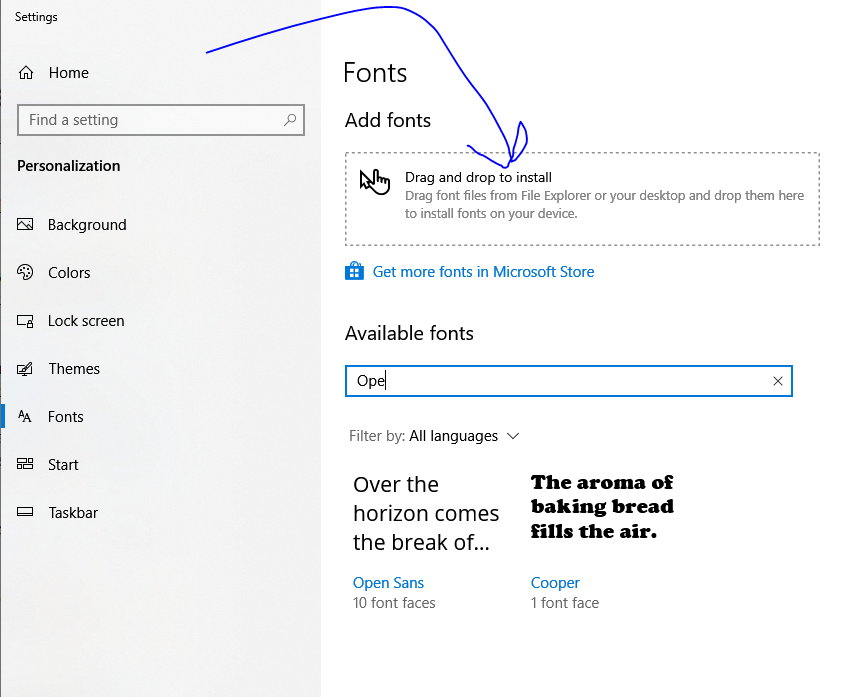
If you want to make changes like add in some text, you aren’t done yet. IPSA uses 3 nonstandard fonts that you probably don’t have installed. At the top of the page there is a button that says “Download fonts”. Do that. Unzip them and then type “Fonts” into your Windows search bar thing.
In Windows 10 (booooooooo!) it’ll look like this.
Predictably, it won’t work quite right, but if you click/drag/drop enough times and say the exact right combination of profanities it will eventually recognize the fonts you uploaded. Powerpoint may not immediately add the new fonts to the font bar, but if you type the name of the font in the box it will update it.
The spectra are annotated in OpenSans. On my screen to match it exactly with a spectra taking up a full slide, my b/y ions are OpenSans 16. This may not be universal. The other 2 fonts are the text around the beautiful spectra.
Why am I adding stuff? I’m just putting in the exact mass of the ions that most clearly illustrate where the NAGs are located in my MS/MS spectra, which is never ever ever on tyrosine.
Now that you’ve got your spectra in, add the right arrows and colors, save the image however you want (probably .TIFF, since the image isn’t compressed.) And then I’m done typing!