….this new manuscript might have the answers to a lot of the questions I think we’ve all been hearing, primarily….
“HOW DO YOU INTEGRATE ALL THIS “-OMICS” DATA!!?!?!”
I’m going with “might have the answers” because there are a lot of assumptions made by the authors regarding the math background of the reader.
When you get to the methods section you get this brief “how we did the proteomics, metabolomics, cyTOF, etc., etc., is all in the Supplemental” and this is the first description of the integration of the data…
….right on….
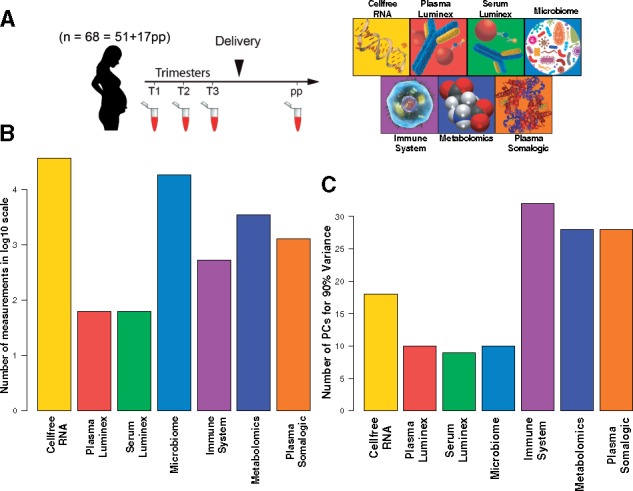
So…all the stuff I’m interested in is in the Supplemental.
The plasma proteomics is done by SomaScan. This is a bead-based array technology that is coming up fast. This one can quantify around 300 proteins per sample. I think we’re going to see it continue to put pressure on LCMS proteomics for a couple of reasons.
1) Biologists are still doing microarrays (for real, they still are) and GWAS. They’ve got all sorts of ways to deal with data that isn’t the most precise thing in the world.
2) Oh — and we still have this reproducibility issue because none of us can agree on a single sample prep method for anything at all, ever.
I really really hope to see a head to head soon to see what the precision/accuracy of this technique is versus someone who is good at proteomics. If anyone sees this, please let me know!
Even at 300 proteins this is still a ton of data across 50+ patient samples and I’m cool with this.
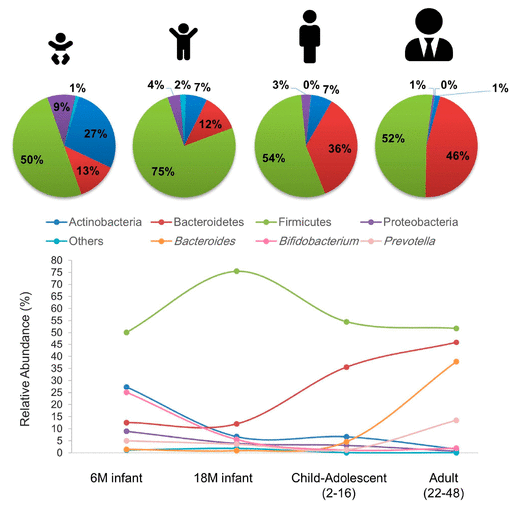
The microbiome stuff was done by a PCR amplification of the 16sRNA and the metabolomics was QE/QE Plus with one running HILIC and the other reversed phase.
The immunomics might be the highlight of the study!
Whole blood from each patient was aliquoted out and either not stimulted or stimulated with LPS or IL-2, and so on — and then cyTOF time! For real, I’ve wondered what the heck you would do with these things (and so have other people, apparently, considering the number I’ve seen sitting around doing nothing after they’ve been purchased). If you aren’t familiar, you essentially put a metal tag on an antibody and then the antibodies bind to the cells and you vaporize everything — I think its inductively coupled plasma, but don’t quote me — then you use the lowest resolution mass spectrometer ever constructed (this home made one made with a spoon might be lower resolution, to be honest). You don’t care, though, because metals differ a lot by mass. (It does limit the total number of antibodies you can use, but it’s still a super cool concept.)

They get a signal, simultaneously, for every cell that comes through their cell sorter thing and gets vaporized, that can provide a concentration of ALL OF THOSE TARGETS! Pretty great, right?
If you’d like to look at the data yourself, it has all been converted to csv and integrated into R. All the scripts are available in a zip file at the very bottom of this page (it says the word “here” in a slightly different color).
Did I learn how to integrate multiomics data? Hmmm…..I’ve got a bunch of math to brush up on that might get me closer than I was before — and — well…I could just use all their scripts and put in my data….so…maybe!