At 3 meetings I spoke at this year, I ran into an enthusiastic person after my talk who wanted to give me Ribo-Seq data so we could compare our proteomes and “translatomes”.
I get a lot of offers to take hard drives full of other people’s data and spend all my free time on it. If you read this blog and can’t relate, you’re probably here for the dog pictures?
You’re welcome! (He’s already bored with this post)
Ribo-Seq (RiboSeq?) is otherwise known as Ribosome Profiling (
wiki). The idea is surprisingly simple. In RNA-Seq (RNASeq?)you measure all the transcripts in a cell. A lot are floating around just doing nothing (probably something but not
correlating directly with protein abundance), but some are with ribosomes making useful proteins. If you dump in an enzyme that chops up RNA all the stuff that is floating around gets chopped up, but the ones at the ribosomes are “protected” and don’t get chopped up.
If you wash away the little chunks of digested DNA (and the enzyme that chopped them up) and liberate the RNA from the ribosome and sequence that then you know what RNA was being translated into protein at that moment.
The test was basically 2 conditions — Relaxed yeast and stressed out yeast (some sort of oxidative stress thing)
Proteomics was performed with a 90 minute gradient on an Orbitrap Fusion II
RNA-Seq and Ribo-Seq were also performed.
The picture at the very top of this post is the correlation. This is very funny to me because earlier this year when I was designing loads of targeted methods for regulated assays I will always remember when a smart young grad student came to me and said — “my R is only 2 9s, is that okay?” That’s when I knew I wasn’t needed there anymore. She was worried about her 7 point SRM standard curve not exceeding 0.99!” Not the same thing, of course, but seeing a RNASeq to proteomics “correlation” of 0.46 puts things into context.
RiboSeq does seem to correspond better to protein abundance. It sure isn’t a 0.9 — even in an organism that only produces around 4,000 proteins under maximum conditions.
Cool study, right?!? A big thanks to the authors because it answers a question a lot of us are going to get. Answer? Yes. It is better. Global proteomics is still better. Targeted protein quan is still the best — if you take proteoforms into account!
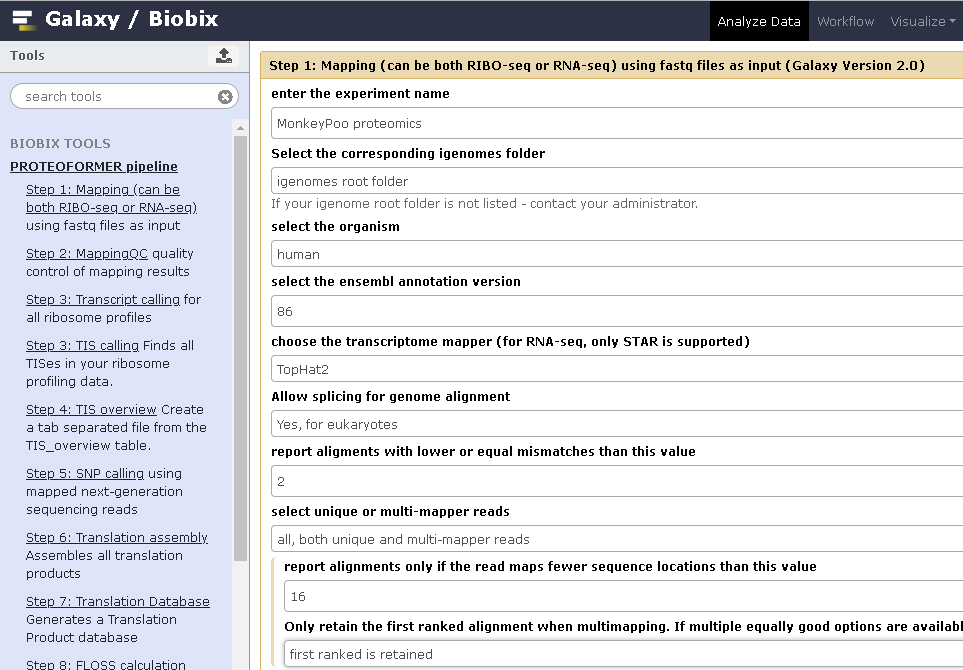
On this topic — I do have some RiboSeq data coming my way in a few months. I fired up the big Linux box and started making a list of pros/cons for changing my Linux distro (I’m running Pop!_OS which is pretty great — natively encrypted, developer centric, but even though it’s built on Ubuntu it isn’t always the most straight-forward for me — the biggest example is that it natively comes with R Commander, which is a step down in my mind from R Studio) because it’s Galaxy and
PROTEOFORMER time!
I don’t know if it was with Proteoformer or the new update, but these great people in Belgium (?) set up a Galaxy instance that we can use! You can link to it from their
github here!