It’s about time that we talked about how to add….
…well…deep learning…(but…come on, I HAD to use that when I found it, right?!?) to your proteomics workflow!
Don’t want to read my rambling about why Prosit is awesome and just want to do it? Skip to Part 2 below!
I almost guarantee that there is someone at your facility who drops all sorts of words like this around — and maybe that same person has given you reason to question their intelligence in other matters, but as long as they keep saying things about “neural networks” and “semi-supervised” whatevers it seems like everyone wants to talk to them, and maybe give them lots of money. Follow this easy walkthough and THAT COULD BE YOU.
I jest, because Prosit is the real deal and has real world advantages, including more and higher confidence identifications right now.
For a biomolecule, the peptide bond is a joy to work with — energetically — crudely optimize the collision energy and you’ll break most of them. Our friends in the small molecule world, where I continue to dabble don’t have it anywhere near as good. There seems to be no rhyme or reason to what energy will break which bonds. When I do QE metabolomics, I step my CE, typically with 10, 30, 100. Just to come close. The ID-X even has something called “assisted” where it tries to help. Most of the time when you’ve got a molecule you really want to study, it makes sense to run it 10 times with different energies….
However — just because peptides are better than most molecules at fragmenting, that doesn’t make them consistent. Look at them. Why on earth would you miss the y7 in this peptide or the y4 in that one? It’s just not there. And — at some level it must make sense –energetically.
Prosit was described here last year:
In as few words as I appear capable of writing — Prosit looks at the ProteomeTools database (you know that thing where they are synthesizing EVERY human peptide and then fragmenting them and making libraries?) and it models the peptides YOU give it against that library with this deep learning thingy.
PART 2: How to use Prosit!
You will need:
1) A protein .FASTA database.
2) The EncyclopeDIA (you can get it here)
3) That’s it. I just felt dumb making a list with 2 entries in it.
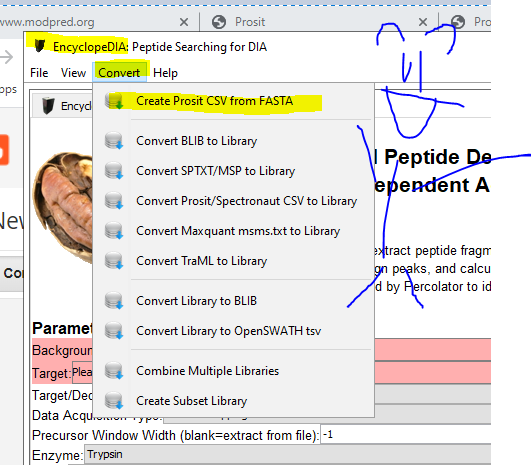
EncyclopeDIA can do all sorts of smart stuff (some of which I wrote not smart stuff about here) — and it also has awesome utilities. Such as “Create Prosit CSV from FASTA”
As an aside, I heard from the Prosit team — they’ll have this integrated soon, but if you wanted to put the words “deep learning” on your ASMS abstract that is due tomorrow you have to do what I am doing.
This is ridiculously easy. Add your FASTA. It will make you a Prosit .CSV file. I believe very strongly in you and your abilities. You’ll definitely be able to do it!
Now — go to proteomicsdb.org/prosit and load that CSV you just made.
Hit next and then tell Prosit the format of your output library:
I’m using MSP because I can’t afford Spectronaut yet. Then submit your job!
Now — this is important. When you submit the job you’ll go into the queue. You’ll want to copy the link URL it gives you and/or the Task ID number. You will not want to close your browser without remembering to do this, because you won’t get your library. When it’s ready you’ll get a download link!
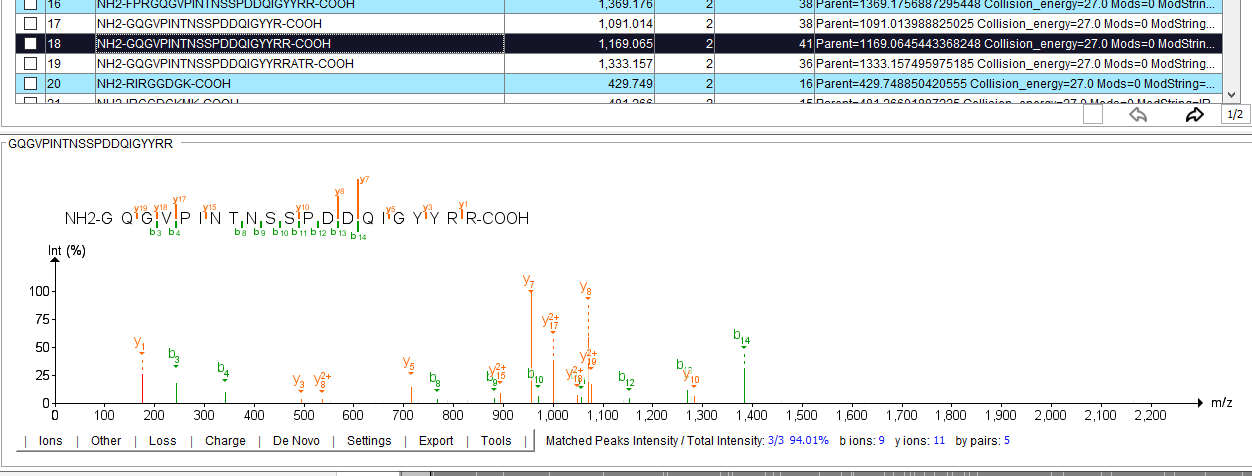
If you want to check the quality of your MSP library — the PDV is a nice, lightweight, java program that will allow you to flip through all of them. If you’ve already got the NIST MS Interpreter installed it will also load them. PDV will look something like this!
For this peptide, Prosit predicts that for a CE of 27 I’m not going to see every b/y ion. There are some bonds that it thinks, from the hundreds of thousands of real peptides it has studied, just won’t fragment well.
And if, for example, you are looking at that real peptide. And it’s right? Then you aren’t penalized for missing that fragment when using this library!