Try forgetting the name of this great new method (you’re welcome! this is the kind of stuff I contribute to society)!
And if it brings some awareness to a fantastic new study that shows some demonstrates a way to fix several of the fundamental problems in my field, then it’s worth it to me if a few more people think I’m an idiot than did yesterday.

I hate phosphoproteomics and I bet you do too. Sure, maybe it pays the bills, but it’s a terrible situation for everyone. The phosphopeptides themselves are finicky. You can’t mess around. If you even lyse the sells just a little bit slower than last time it seems like things have changed. And I don’t care what kit you’re looking at. You’ll see 3 barely visible beads in the lid of the tubes you bought and you’ll think, just for a second, about maybe calling out sick the rest of your career. And even when you (or someone way better at sample prep than you, in my case) does everything perfect the replicates are often still sad. There are too many variables — there is too much sample handling. There are too many steps. There are too many places where a pH off by 0.1 will ruin the number of those awful looking fragmentation spectra that you’re going to get in the end.
Would someone please just find an affordable robot and do the most incredibly boring study in the history of the galaxy and just work out every miserable variable and just provide a realistic way to do large scale phosphoproteomics prep??? That’s all I’m asking for. Oh — and please make it open access. I’m off campus….
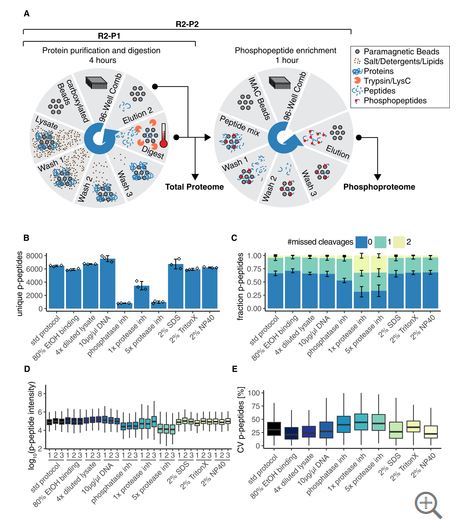
How does R2-P2 do?
Cheap Robot? BOOM!
KingFisher!! (Not OpenTrons cheap, but affordable as robots go [and, btw, if you didn’t know, it is marketed by multiple companies under different names with different lables on it. Shop around, you can get them 1/2 or 1/3 price depending on if it says RioBad or FermoTisher or something else (I forget the third one). Not making that up.
All the boring details worked out? BOOM!
This group did everything. I respect and pity them for the amount of work this study must have been (I guess it got easier once the robot was functional)
Compatible with large scale?
BOOM!
Maybe yeast phosphoproteomics isn’t the most complex system, but they do a bunch of replicates and perturb some central MAPK systems and the data looks awesome. It seems very realistic that you could scale this up to larger systems easily.
And the paper is open access!
Look — this isn’t the first automated phosphoproteomics prep system, and it won’t be the last, but this checks all the right boxes. 100% recommended paper.
You’re fragmentation spectra will still look like garbage, but that’s physics and chemistry and stuff — what we need is a reproducible way of getting to the same garbage spectra every time.